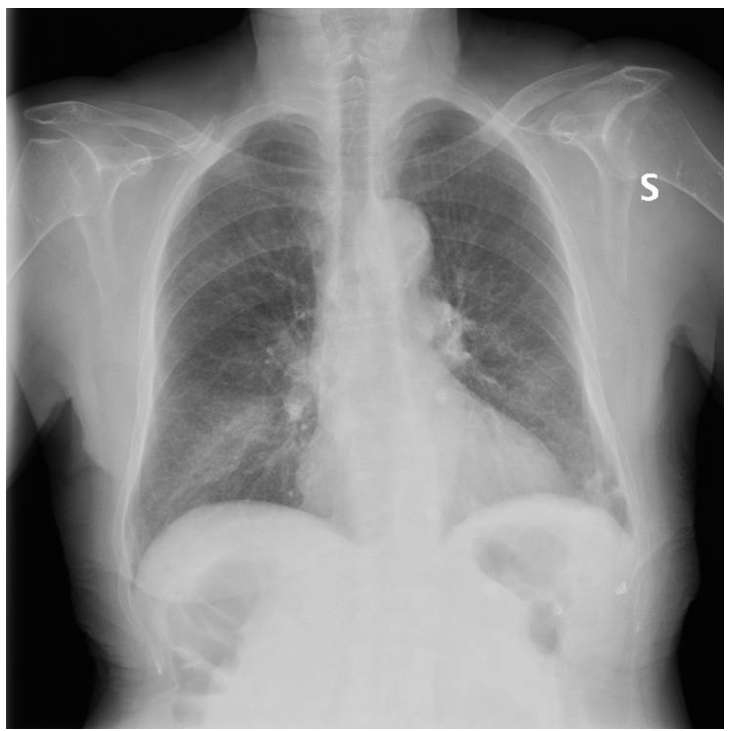
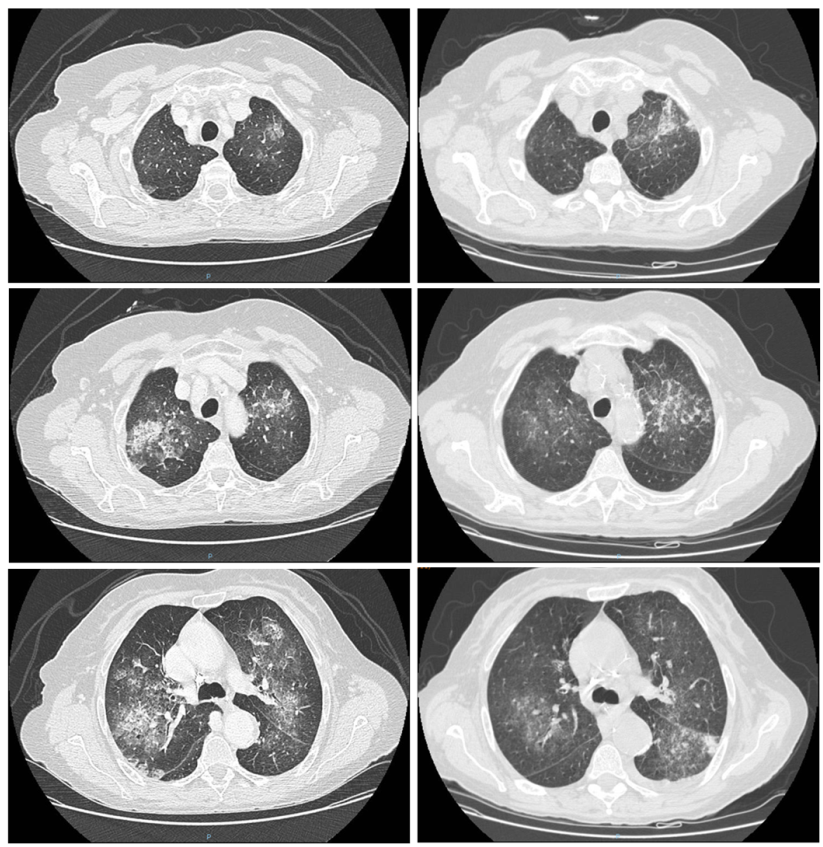
Raisonnement clinique pour établir un diagnostic
Le tableau clinique soulève la question du diagnostic différentiel d’une pneumonie associée à une hyperéosinophilie. Les principales causes à considérer dans ce contexte sont listées ci-dessous.
- Infections à helminthes : les principaux agents responsables sont Ascaris (A. lumbricoides et A. suum), Ancylostoma duodenale, Necator americanus, Toxocara canis, Trichinella, Schistosoma et Strongyloides stercoralis. Notre patiente ne rapporte aucun antécédent de voyage à l’étranger ces dernières années. Par ailleurs, il n’existe aucun antécédent de contact avec des animaux domestiques ou sauvages, ni de risque d’exposition à une infestation à Strongyloides (la patiente ne possède pas de potager et ne fréquente pas d’environnements sauvages). Néanmoins, une recherche directe de parasites dans les selles et les sécrétions respiratoires a été effectuée, qui s'est révélée négative, tout comme la sérologie pour Strongyloides stercoralis.
- Réaction aux médicaments et toxines : le spectre des manifestations iatrogènes peut aller d’un simple épaississement asymptomatique à des conditions potentiellement fatales telles que le DRESS (drug reaction with eosinophilia and systemic symptoms). Cette hypothèse n’est pas soutenue chez cette patiente, dont l’historique thérapeutique est essentiellement inchangé, à l’exception des corticoïdes inhalés, utilisés depuis des années.
- Pneumonie idiopathique aiguë : survient habituellement après le début ou la reprise du tabagisme ; elle peut également résulter d’une exposition aiguë à des poussières ou vapeurs chimiques.
- Pneumonie éosinophilique chronique : touche principalement les femmes non-fumeuses, comme dans ce cas, mais implique habituellement une atteinte surtout sous-pleurale, très différente de celle observée chez la patiente. Elle peut être associée à une radiothérapie antérieure pour cancer du sein.
- Aspergillose bronchopulmonaire allergique : réaction d’hypersensibilité complexe due à l’exposition à Aspergillus. Elle se manifeste habituellement sous forme d’asthme chronique, avec exacerbations fréquentes. Elle est associée à fièvre, malaise, hémoptysie et éosinophilie périphérique. Toutefois, dans ce cas, le scanner thoracique montre un schéma différent, avec prédominance de bronchiectasies touchant principalement les apex.
- Syndromes hyperéosinophiliques : syndromes paranéoplasiques, réactifs ou idiopathiques, caractérisés par une hyperéosinophilie (> 1 500 cellules/µl) et une atteinte potentielle d’autres organes (poumons, cœur, cerveau, tube digestif, reins). Les manifestations cliniques sont dues à l’infiltration tissulaire par les éosinophiles. Le diagnostic est posé en présence de manifestations cliniques compatibles et d’une hyperéosinophilie, après exclusion des autres conditions pouvant expliquer le tableau clinique.
- Granulomatose éosinophilique avec polyangéite (GEPA, anciennement syndrome de Churg-Strauss) : vascularite associée aux ANCA, caractérisée par la présence de sinusite, d’asthme et d’hyperéosinophilie périphérique. Radiologiquement, elle se caractérise par des opacités pulmonaires transitoires.
Compte tenu de la suspicion clinique de granulomatose éosinophilique avec polyangéite, la patiente a subi des examens complémentaires :
- Évaluation ORL : du côté droit, la muqueuse septale et le cornet inférieur semblaient atteints par des lésions suspectes d’origine vasculitique ;
- Test P-ANCA, positif à un titre élevé (186 UI/ml).
Granulomatose éosinophilique avec polyangéite (GEPA)
La granulomatose éosinophilique avec polyangéite (GEPA), anciennement appelée syndrome de Churg-Strauss, est une vascularite nécrosante systémique touchant les petits et moyens vaisseaux sanguins. Elle se caractérise par une éosinophilie périphérique, de l’asthme, une rhinosinusite avec ou sans polypes, et une atteinte d’autres organes.
La GEPA est classée parmi les vascularites systémiques dites associées aux ANCA (anti-neutrophil cytoplasmic autoantibodies), bien que la positivité des ANCA ne soit présente que chez 30 à 40 % des patients. Les ANCA, généralement de type p-ANCA (myéloperoxydase), permettent de distinguer deux sous-groupes au sein de la GEPA.
Les patients GEPA ANCA-positifs présentent fréquemment des manifestations telles que multinévrite, glomérulonéphrite extracapillaire et purpura cutané. En revanche, les patients GEPA ANCA-négatifs présentent plus souvent des infiltrats pulmonaires, une polypose nasale, une myocardite et une pleurésie. D’autres manifestations cliniques, telles que symptômes systémiques (fièvre, asthénie, arthromyalgies, perte de poids), se distribuent de manière similaire dans les deux sous-types. La GEPA est une maladie rare, présentant l’une des incidences et prévalences les plus basses parmi les vascularites systémiques.
En Europe, les données du Groupe français d'étude des vascularites montrent une prévalence comprise entre 10,7 et 13 cas par million d’habitants, avec une incidence de 0,5 à 0,8 nouveaux cas par an et par million d’habitants. L’apparition peut survenir à tout âge, mais elle est plus fréquente chez l’adulte, avec un âge moyen d’environ 50 ans. Les cas pédiatriques sont très rares.
L’étiologie de la GEPA demeure inconnue. Les causes de l’inflammation éosinophilique et de l’activation incontrôlée du système immunitaire avec tempête cytokinique restent indéterminées.
Les critères de classification ont été révisés en 2022 par l’American College of Rheumatology / European Alliance of Associations for Rheumatology (EULAR) et sont les suivants :
- asthme (+ 3 points)
- polypes nasaux (+ 3 points)
- multinévrite ou polyneuropathie (+ 1 point)
- éosinophilie > 10 % dans le sang périphérique (5 points)
- preuve histologique de vascularite avec éosinophiles extravasculaires (+ 2 points)
- cANCA élevés (anticorps anticytoplasme des polynucléaires neutrophiles) ou anticorps anti-protéinase 3 (– 3 points)
- hématurie (– 1 point).
Un score ≥ 6 a une sensibilité de 85 % et une spécificité de 99 %.
Évolution clinique et issue
À la lumière du tableau clinique, un diagnostic de GEPA a été retenu. La patiente présentait un asthme, une hyperéosinophilie, des infiltrats pulmonaires transitoires et migrateurs, et une atteinte ORL (la patiente a rapporté une sinusite chronique à l’interrogatoire). En l’absence de facteurs pronostiques négatifs, une corticothérapie a été initiée à la dose de 1 mg/kg/jour de prednisone, associée secondairement au méthotrexate.
Dès la première administration de corticoïdes, une amélioration marquée des paramètres biologiques a été observée, avec une disparition complète de l’éosinophilie, restée négative 3 mois après le début du traitement. De plus, une amélioration progressive des symptômes respiratoires a été notée, avec résolution de l’insuffisance respiratoire.
La patiente est sortie après 13 jours d’hospitalisation. Par ailleurs, après le début d’une thérapie martiale, une normalisation progressive des valeurs d’hémoglobine a été observée. L’anémie a été attribuée à des pertes chroniques liées aux polypes intestinaux, en association avec une composante probablement imputable au trouble chronique sous-jacent.
Sources
- Bellan M, Delsignore E, Manfrinato C, Critto O, Barasolo G, Francese M, Olivetto L, Terribile R, Bertoncelli MC. Un caso di polmonite associata a ipereosinofilia. Federazione delle Associazioni dei Dirigenti Ospedalieri Internisti. 01 feb 2018
- Solans-Laqué R, Rúa-Figueroa I, Blanco Aparicio M, García Moguel I, Blanco R, Pérez Grimaldi F, Noblejas Mozo A, Labrador Horrillo M, Álvaro-Gracia JM, Domingo Ribas C, Espigol-Frigolé G, Sánchez-Toril López F, Ortiz Sanjuán FM, Arismendi E, Cid MC. Red flags for clinical suspicion of eosinophilic granulomatosis with polyangiitis (EGPA). Eur J Intern Med. 2024 Oct;128:45-52. doi: 10.1016/j.ejim.2024.06.008. Epub 2024 Jun 15. PMID: 38880725.
- Emmi G, Bettiol A, Gelain E, Bajema IM, Berti A, Burns S, Cid MC, Cohen Tervaert JW, Cottin V, Durante E, Holle JU, Mahr AD, Del Pero MM, Marvisi C, Mills J, Moiseev S, Moosig F, Mukhtyar C, Neumann T, Olivotto I, Salvarani C, Seeliger B, Sinico RA, Taillé C, Terrier B, Venhoff N, Bertsias G, Guillevin L, Jayne DRW, Vaglio A. Evidence-Based Guideline for the diagnosis and management of eosinophilic granulomatosis with polyangiitis. Nat Rev Rheumatol. 2023 Jun;19(6):378-393. doi: 10.1038/s41584-023-00958-w. Epub 2023 May 9. PMID: 37161084.
- Chung SA, Langford CA, Maz M, Abril A, Gorelik M, Guyatt G, Archer AM, Conn DL, Full KA, Grayson PC, Ibarra MF, Imundo LF, Kim S, Merkel PA, Rhee RL, Seo P, Stone JH, Sule S, Sundel RP, Vitobaldi OI, Warner A, Byram K, Dua AB, Husainat N, James KE, Kalot MA, Lin YC, Springer JM, Turgunbaev M, Villa-Forte A, Turner AS, Mustafa RA. 2021 American College of Rheumatology / Vasculitis Foundation Guideline for the Management of Antineutrophil Cytoplasmic Antibody-Associated Vasculitis. Arthritis Rheumatol. 2021 Aug;73(8):1366-1383. doi: 10.1002/art.41773. Epub 2021 Jul 8. PMID: 34235894.
- Pyo JY, Lee LE, Park YB, Lee SW. Comparison of the 2022 ACR/EULAR Classification Criteria for Antineutrophil Cytoplasmic Antibody-Associated Vasculitis with Previous Criteria. Yonsei Med J. 2023 Jan;64(1):11-17. doi: 10.3349/ymj.2022.0435. PMID: 36579374; PMCID: PMC9826961.